As a child, Samuel Idahosa was accustomed to frequent, debilitating pains. He didn’t know he had sickle cell anemia – a diagnosis his parents opted against disclosing to him due to the social stigma in Nigeria, where they lived.
Idahosa finally learned the truth about his condition not long after his family moved to California in 2016. He continued to endure regular hospitalizations, although his pain was better managed than it had been in Nigeria.
That’s now changed, however: After undergoing a novel gene therapy treatment in 2025 at UCLA Health, Idahosa, 24, is considered cured.
Idahosa was honored recently at a UCLA men's volleyball team practice at Pauley Pavilion.
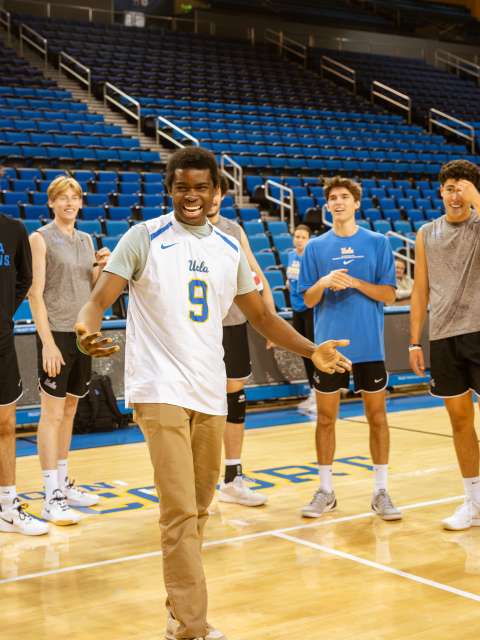
Living with sickle cell in Nigeria
Nigeria has long had one of the highest rates of sickle cell disease. Together with several other countries in sub-Saharan Africa, it accounts for about 80% of worldwide cases, according to the World Health Organization. In the United States, sickle cell is the most common genetic blood disorder and likely affects more than 100,000 people, the U.S. Centers for Disease Control and Prevention estimates.
Sickle cell disease causes the red blood cells to be misshapen into a crescent, or sickle, shape that restricts regular blood flow. The sickled red blood cells are stickier than normal and clump together, leading to bouts of excruciating pain. The cells also have a shorter life span, which results in chronic anemia due to a shortage of red blood cells.
Sickle cell occurs when a person inherits the gene from both parents. Someone can be a carrier of the gene and not have any symptoms but can still pass the gene to their children. Because of this, Idahosa’s parents both underwent mandatory premarital testing in Nigeria to see if they were carriers. His mother was identified as a carrier, but his father was not – an error that wasn’t discovered until after Samuel began exhibiting symptoms and his father got retested.
“Growing up, it was really hard,” Idahosa said. “Other kids could do whatever they wanted but for me it was stricter, because any little thing could send me to the hospital.”
While there were other kids in school who had been identified as having sickle cell and weren’t allowed to do many activities, including sports, they were also frequently pitied or even mocked back then, Idahosa recalled.
“My parents didn’t tell me,” he noted, “but they gave me really strict guidelines.”
Even outside of school, Idahosa was forbidden from joining other kids engaged in physical activities. “I was just staying at home, watching the kids and wishing I could do that,” he said. “I couldn’t play sports; I couldn’t run. I played hide-and-seek, but I was always hiding and not running.”
Eventually, though, he rebelled against the restrictions, running out on the field at school and careening wildly so his friends would chase him, despite knowing he’d quickly come close to passing out.
“It just got to the point,” he said, “where I didn’t care.”
The consequences were severe: Idahosa began to be hospitalized at least once a month. During each one-to-two-week stay, he received an intravenous saline solution to help guard against dehydration and increase blood volume. He also received pain medications, but they were only partially effective.
“It was hell,” Idahosa said.
Back then, the pain was always in his stomach. Idahosa developed a high pain threshold, he said, and learned how to determine when it was unbearable enough to require hospitalization.
“There’s a breaking point,” he explained. Pain that didn’t require hospitalization was akin to a severe migraine lasting several days, he said, but if it got to the point where he was holding his breath to control the pain, he knew he’d have to be admitted for care.
By the time he was about 14, he was no longer attending school, having completed all of his required work at home. It was a narrowly prescribed existence: “I was just at home resting and not doing anything that might trigger it.”
A move to the US for better treatment
Following his family’s 2016 move to California when he was 15, Idahosa was initially able to avoid triggering a pain crisis, largely because his activities were limited. Although he was back in school, “those first years, I was home a lot,” he said.
During this time, Idahosa had regular medical checkups. But it wasn’t until a blood test at age 16 that he finally learned he had sickle cell.
“I was surprised, because I knew what it was, but had always been told I didn’t have it,” he said. He also finally learned that his condition had been the main driving force behind the family’s move, and that the death of his younger sister in Nigeria was due to sickle cell.
Idahosa began taking hydroxyurea, a medication approved by the Food and Drug Administration in 2017 for use in children to reduce the symptoms of sickle cell.
However, as Idahosa’s social circle and activities increased, his painful episodes, which had been occurring every other year, became more frequent. A trip to Idyllwild in the nearby San Jacinto mountains caused a particularly painful crisis, given that lower oxygen levels at higher altitudes place additional demands on the cardiovascular system.
By this point, Idahosa was being monitored monthly and was receiving additional newly FDA-approved oral and intravenous medications to help reduce the frequency and intensity of his pain crises.
They weren’t always enough to stave off hospitalization, however.
Newly approved cure
Up until recently, the only sickle cell cure has been a bone marrow transplant, generally from a close relative who is a full match. This presents the first hurdle: often, siblings or other close relatives may only be a partial match. These “half-match” transplants require high-dose chemotherapy and carry additional potential risks for complications.
Idahosa’s youngest brother, who doesn’t have sickle cell, was only a partial match for him. (He was, however, a full match for their other brother, who had sickle cell but with much milder symptoms, and was able to undergo a bone marrow transplant in 2024.)
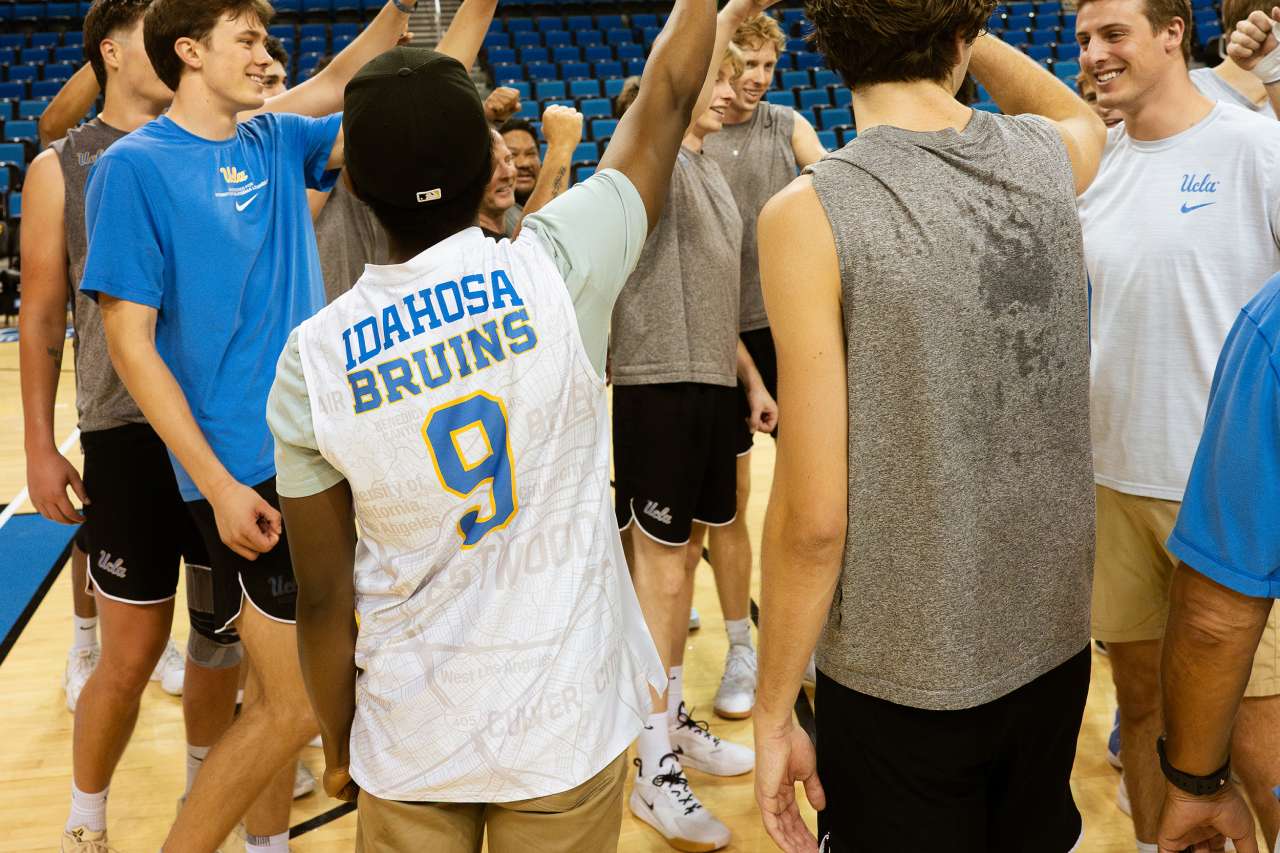
In late 2023, the FDA approved two new gene therapies to treat sickle cell disease. Not long after, UCLA Health became one of the first sites to offer one of the treatments, Lyfgenia, which extracts and then modifies the patient’s own blood cells.
“It changes the DNA blueprint of these stem cells by inserting a gene for modified hemoglobin,” explained Gary J. Schiller, MD, clinical chief of the UCLA Health Sickle Cell Disease Program and a professor of medicine at the David Geffen School of Medicine at UCLA. “This allows those stem cells that have been successfully altered with the addition of the gene to produce a hemoglobin that does not sickle,” which allows them to produce new healthy red blood cells.
These genetically altered blood cells are likely to out-survive the person’s native blood cells, he said, so that about four to six months after the treatment, the modified cells comprise at least half of the red blood cells circulating in the body.
Undergoing gene therapy
Idahosa, who enrolled at UCLA as an undergraduate transfer student in 2023, moved his health care to UCLA Health at the same time. He soon learned about the Lyfgenia treatment from a professor who knew he had the disease and from one of the nurse practitioners who was treating him.
In June of 2025, Idahosa graduated from UCLA with a degree in molecular, cell and developmental biology. In July, he was admitted to Ronald Reagan UCLA Medical Center to undergo Lyfgenia therapy.
In preparation, Idahosa was put on an exchange transfusion protocol to decrease the percentage of cells with hemoglobin S, the type of hemoglobin that causes sickle cell.
“The red blood cells are sequentially exchanged through a procedure called hemapheresis,” explained Dr Schiller, a member of the UCLA Health Jonsson Comprehensive Cancer Center. “We initiate that first, repeating it monthly, to get the percentage of hemoglobin S low.”
The cells were extracted via a catheter placed near the groin. Each session lasted several hours, Idahosa recalled. “It just felt weird,” he said, “like a tingling sensation, but with a lot of pressure.”
Idahosa was subsequently injected with a drug to induce the stem cells to exit the bone marrow and enter his bloodstream, then was hooked up to a centrifuge machine so the various blood components could be separated by weight.
“We can capture them on the same machine we use for the red cell exchange,” Dr. Schiller explained, “but in this case, we’re collecting all kinds of white cells in the blood, a percentage of which will be the stem cells that were mobilized to leave the marrow.”
A small portion of Idahosa’s stem cells were retained and sent to an outside laboratory so the gene necessary for producing normal hemoglobin could be added.
Once the cells were ready, Idahosa was hospitalized in preparation for having them infused. He received daily chemotherapy for four days to eliminate the sickled cells and prepare his bone marrow to receive the newly modified cells. Two days later, the cells were injected into his blood stream.
“These cells know where to go. You put them in the blood, and they’ll be in the bone marrow in 10 minutes,” Dr. Schiller said.
Idahosa still needed to remain in the hospital, however. “Even after the cells are administered, the chemotherapy induces low white count, which puts the patient at risk for infection; low hemoglobin, which puts them at risk for anemia; and low platelets that put them at risk for bleeding,” Dr. Schiller explained. Idahosa was closely monitored and received various transfusions and medications to counteract these, he said.
Patients’ blood counts generally begin to recover in about two to four weeks, Dr. Schiller said, allowing them to be discharged.
During this time, Idahosa was also experiencing side effects from the chemotherapy, including difficulty swallowing and an intensely sore throat. “Just swallowing my saliva was so painful,” he said.
The long hospitalization was difficult, he acknowledged: “I went from freedom to being stuck in a room for a whole month.”
Dramatic improvement
At first Idahosa returned to UCLA Health once a week for monitoring. That’s now tapered off, although his hemoglobin levels will continue to be assessed every six to eight weeks for the first year, Dr. Schiller said.
A few months after the treatment, Idahosa felt the familiar beginnings of stomach pain. This time, however, he could tell immediately that it wasn’t severe enough to require hospitalization. Most telling, he hasn’t needed any pain medication for several months.
Idahosa has had some residual sickle cell pain in his left arm, he noted, but it’s nothing like before.
His hemoglobin levels have increased and are now only slightly below what’s considered normal. “I’m really improving,” he noted.
He’s currently receiving all of his standard vaccinations again, given that the chemotherapy significantly reduced his previous levels of immunity. Until then, his outings are mostly limited.
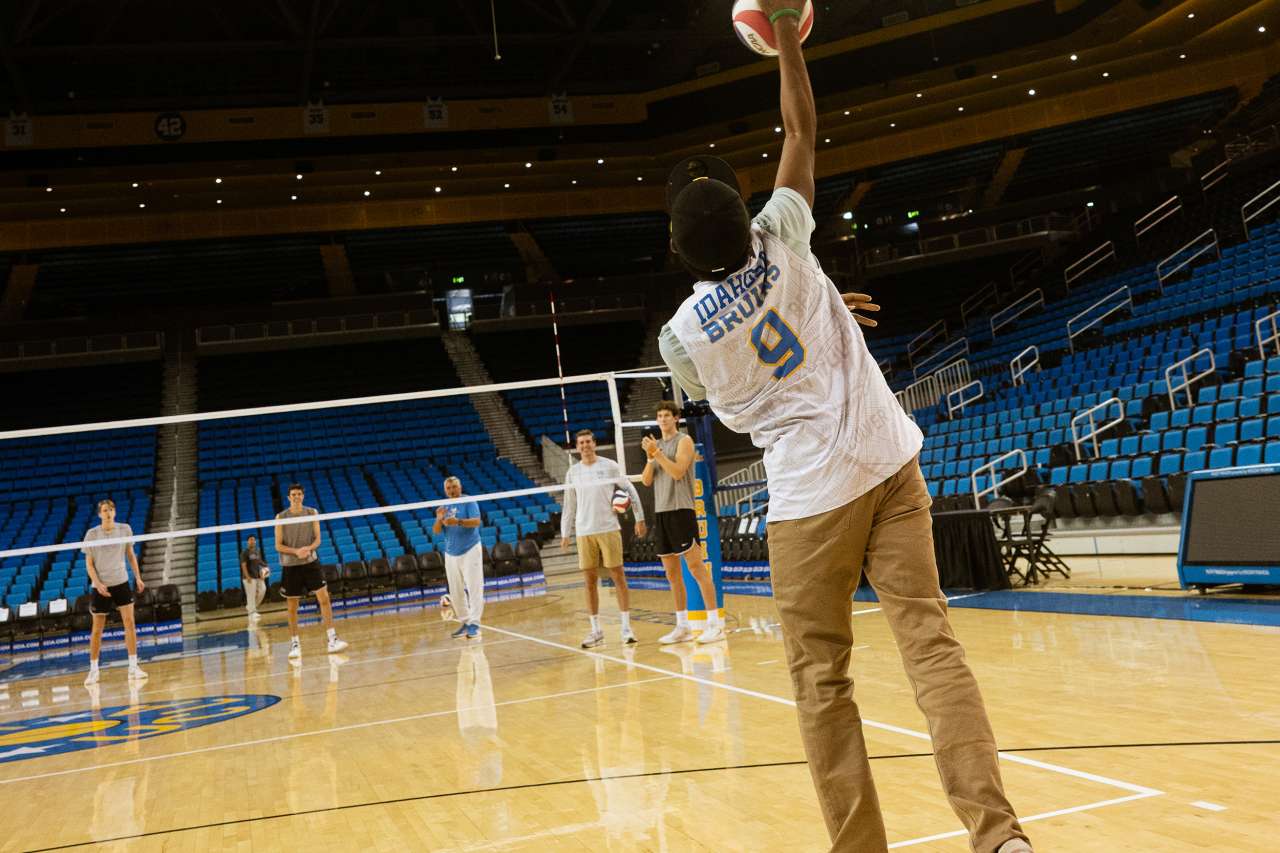
Idahosa looks forward to being able to play volleyball himself and also plans to try swimming and scheduling another higher-altitude excursion. Longer term, he hopes to attend medical school and eventually return to Nigeria to help others with sickle cell.
A life-changing treatment
“We’ve had excellent results in everybody who’s gone through the gene therapy,” Dr. Schiller said.
That said, “this will not be for everybody,” he noted. “What we’ve discovered is that patients who are on chronic high doses of narcotics have marginal benefit from gene therapy. They may have already sustained so much damage to their blood vessels, or they’ve suffered the effects of prolonged and intense narcotic opiate therapy, that they don’t necessarily benefit.”
Still, for those who have undergone the Lyfgenia treatment, sickle cell no longer overshadows their lives. “There are very few treatments that have such a dramatic response,” Dr. Schiller said.